


























 For Chemical Sciences With a Grant of Rs. 60 Lakhs")






An Introduction To Cheminformatics And Drug Discovery
Cheminformatics is the study of all aspects of the representation and use of chemical and related biological information on computers. It has applications in drug discovery health, data mining, and many other areas. According to the National Center of Biotechnology Information, cheminformatics is a relatively new field of information technology that focuses on the collection, storage, analysis, and manipulation of chemical data. The chemical data of interest typically include information on properties, spectra, small molecule formulas, structures, and activity (biological or industrial).
Cheminformatics originally emerged as a channel to assist the discovery and development process of drugs. However, cheminformatics now plays a crucial role in many areas of chemistry, biochemistry, biology. Since the 1950s, several foundational algorithms of cheminformatics have been described. But open-source software, implementing the algorithms, became accessible only since the mid-1990s, which is around 40 years later. But in 2004, a large public small molecule structure repository was made freely available by the National Library of Medicine.
Bridging Cheminformatics And Computer Science
Let’s look at the relationship between computer science and cheminformatics to understand how computer science contributes to cheminformatics. Just how Bioinformatics talks about the genetic sequence, cheminformatics deals
with the chemical structures. Cheminformatics is used mostly to reduce the risk during drug discovery by minimizing the chances of small molecules failing due to poor physical, chemical, or biological properties during the development of a drug candidate. In cases where these properties are poor, a cheminformatics approach can suggest the replacement of certain functional groups to maintain the potency but improve the solubility and bioavailability. This can reduce the cost of drug discovery by eliminating the need for trial and error method.Many molecules exist in this world. A 2D or a 3D structure can represent all these molecules. Although the 2D or 3D structure gives different pieces of information, both are valid and accurate. All this information generates an enormous amount of data. Although computer science can help us to store all these, there are a few challenges in integrating these data.
There are many databases available online; two of those are ChEMBLdb and PubChem, which are freely available and contain possibly millions (ChEMBLdb) and tens of millions (PubChem) of data points. But, it is hard to normalize and standardize since different databases use different algorithms. The sheer data volume and the difficulties in the normalization of chemical and bioactivity data make the integration of data across chemical databases, a huge challenge. Due to this, we cannot convert data from one database to another database, making it hard to normalize.
Also, the molecular draft must be captured in a machine-readable fashion. The input we put should be readable by the machine so that the outcome we get is an excellent 3D or 2D structure, which we want to share with other scientists for their future research. It is also necessary that life scientists are able to search for chemical data like multi-labeled graphs whose graph labels can change from database to database. So when you change from one database to another database, the label should stay intact, and you should get exact data of the 2D or 3D structure of the molecule.
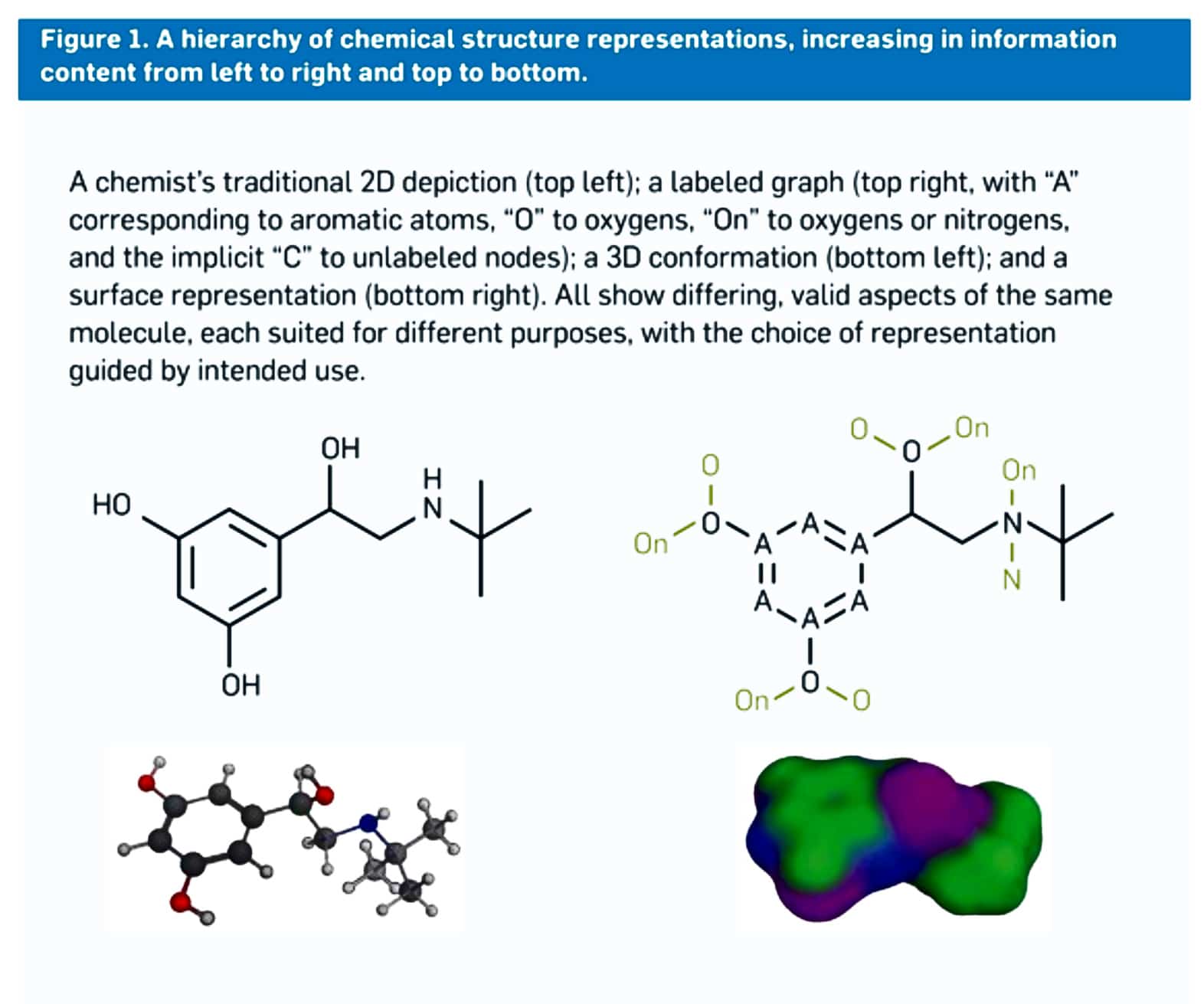
Figure 1: shows that there are different ways or many ways to represent the structure of a molecule. For example, on the top left, it’s a 2D structure where it’s not fully labeled, and on the top right, it’s fully labeled 2D structure. It’s a 3D confirmation on the bottom left, and a surface representation on the bottom right. So there are four ways to represent a molecule. Each of those gives different pieces of information. But all four are accurate and valid.
There are many databases online, and different databases use different algorithms. So it has been hard to normalize all of them, and there are too many molecular structures that make it difficult for scientists to search for a specific molecule structure. This problem was solved by the canonicalized algorithm and identifier. Morgan developed the first canonicalization algorithm or the ‘unique representation algorithm’, allowing chemists to create a unique string representation of the chemical graph and use the string comparisons to compare structures. Later in 1988, Weininger defined a Simplified Molecular-Input Line-Entry System, also known as the SMILES format.
Multiple implementations of the format became available as the original canonicalization algorithm was proprietary, each employing a different canonicalization algorithm, usually based on the morgan algorithm. As the amount of data available online kept on increasing, the use of unique structure-based identifiers became an urgent need, resulting in the development of a non-proprietary, structured, textual identifier for chemical entities called the International Union of Pure and Applied Chemistry’s International Chemical Identifier (InChI). With this InChI code, it has been easy for all the scientists to search for a specific molecule structure where each of the molecule structures or molecules has its own InChI code.
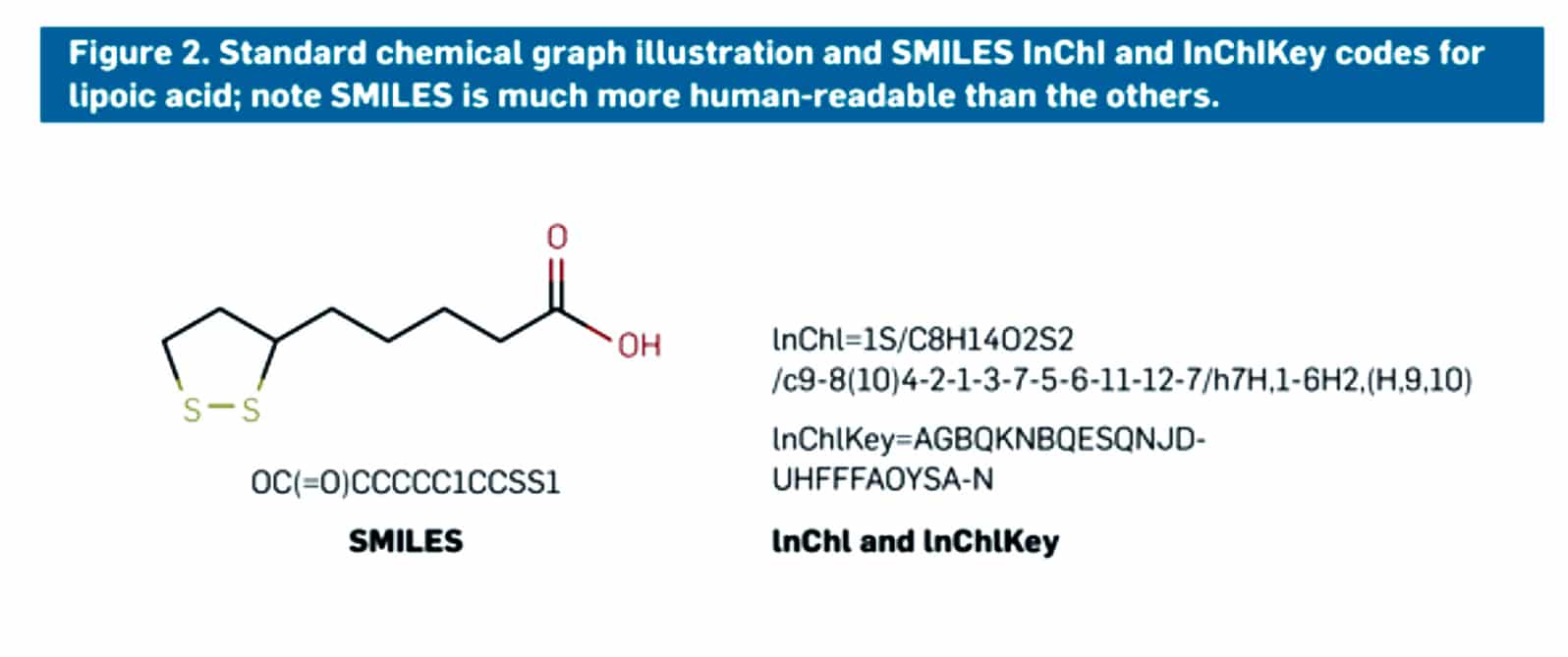
Figure 2: shows a molecule of lipoic acid with the two types of codes that made scientists easily identify a molecule or molecular structure. A SMILES code is given at the bottom left as described earlier, and on the right side, the InChI and InChIKey codes are mentioned. The codes make it easy for everyone to access the website and databases and easily obtain the information or data for research purposes or educational purposes.
Activity Mining And Prediction
Why cheminformatics is important and how cheminformatics can contribute to science?
The answer is cheminformatics allow prediction, which decreases errors. The objective of any modeling approach is to capture and characterize the relationship between structural features and observed biological activity. Rather than designing a drug in a lab that may cost more and might be prone to failure, we can just do that in a computer which gives us the structural features and observed biological activities. Using this approach, we can predict the activity as well as the weakness of a drug to improvise it further.
How is Chemoinformatics Used in Drug Discovery?
The normal process of drug discovery involves searching for potential molecules and compounds that can be used to minimize the severity of a particular disease. Several stages of screening are done to compare the effectiveness of these potential molecules to stop a biochemical mechanism.
This process can be drastically accelerated with the help of cheminformatics, whose principal application in research is the discovery and development of drugs. There are many techniques, software, and resources available to achieve this. Let’s look at some of the applications of cheminformatics in drug discovery.
- Databases of relevant compounds and metadata
Chemical databases of compounds and molecules relevant to drug discoveries are the backbones of computer-aided drug discovery, whether it is chemoinformatics or bioinformatics. These databases provide information that can be utilized to build knowledge-based models for designing and discovering drug molecules. Most popular databases include PubChem, ChEMBL, ZINC, and NCI.
- 2D virtual screening
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules so that those structures which are most likely to bind to a drug target, typically an enzyme or protein receptor can be identified. In 2D virtual screening, databases of chemical structures are interrogated to find compounds that are similar to known actives (similarity searching).
- 3D virtual screening
In 3D virtual screening, databases of chemical structures are interrogated to find compounds that possess a pharmacophore or substructure in common with a known active (pharmacophore and substructure searching).
- Property prediction
Predicting properties like solubility and logP is very important in pharmacology and drug discovery.
- ADMET prediction
It is important to understand the chemical and physical characteristics of drug molecules in order to predict how exactly a drug molecule is absorbed by the body, distributed around the body, metabolized, and excreted. It is vital to know the toxicity of the drug in these circumstances. Such studies are referred to as ADMET.
- Clustering
Clustering of chemical compounds, otherwise known as unsupervised machine learning is used in drug discovery, mostly in preliminary analyses of large data sets of medium and high dimensionality as a method of selection, diversity analysis, and data reduction. Compared to the other costs of drug discovery, clustering can add significant value at a minimal cost.
- Quantitative structure-activity relationship modeling
Quantitative structure-activity relationship (QSAR) is a computational modeling method for revealing relationships between structural properties of chemical compounds and biological activities. QSAR modeling is essential for drug discovery as it predicts how functionalizing the compound relative to a chemical series can increase its potency and other properties.
- Modeling or predicting protein-ligand binding
Identifying possible ligand-enzyme interactions is of major importance in many drug discovery processes as enzymes are one of the most important groups of drug targets. There are computational methods that can apply the information from resolved and available ligand-enzyme complexes to model new unknown interactions and therefore contribute to answering open questions in the field of drug discovery like the identification of off-target binding, unknown protein functions, induced-fit simulations, and ligand 3D homology modeling.
- De novo design
It involves designing molecules from scratch. De novo drug design is an iterative process in which the 3D structure of the receptor is used to design novel drug molecules. novo designing, the structure of the lead target complexes are determined and lead modifications are designed using molecular modeling tools.
Cheminformatics is a very useful field of science that highly contributes to science. It is extremely helpful in designing and discovering drugs to avoid the try and error method, which might be very costly. Cheminformatics also made it easy for scientists to search for molecules, their structures, and detailed information using the numerous databases available. Therefore, a focused and in-depth study in cheminformatics could help scientists and professionals in developing new drugs for many diseases that are still a threat to humanity.

For those students and professionals who wish to enter the field of cheminformatics and contribute to drug discovery, Rasayanika is offering an online certification course in Cheminformatics & Drug Discovery.
The course is intended to provide you with a strong foundation in cheminformatics and drug discovery by covering topics like Computer-Aided Drug Design, Tools used for chemo-informatics, In silico generation of virtual molecules, etc.
Cheminformatics Career
A course in Chemoinformatics can help you –
- To develop methods for performing statistical analysis of large datasets.
- To develop methods for making and storing data on molecular structures, reaction mechanisms, molecular interactions, etc.
- To work as researchers in the laboratory.
- It will even help you to identify chemical properties and trends from large databases.
After completing the chemoinformatics course you have the opportunity to work in fields like-
- Bioinformatics
- Computational chemistry
- Materials science
- Pharmaceutical research
- Theoretical chemistry
The job profiles which you can apply for on the basis of this course are-
- Chemoinformatics scientist
- Computational chemist
- Chemical data scientist
- Information analyst
- Information officer
- QSAR software tester
- Technical editor
- Research assistant
Some institutions offer degree programs in cheminformatics, including bachelor’s, master’s degrees, and Ph.D. Cheminformatics specialists should gain a strong background in chemical structures and principles, as well as a combination of knowledge from both computer science and mathematical background.
A Ph.D. is necessary if you want to advance in your career. A postdoctoral degree can also be useful in developing advanced skills or in making the transition from another chemical specialty.
Keywords: cheminformatics career, cheminformatics online course, cheminformatics certification course, cheminformatics & drug discovery, cheminformatics tools, cheminformatics introduction.












